Cell type mapping benchmark
Last compiled: 20 March 2026
cell_type_mapping_benchmark.RmdLoad required packages
library(semla)
if (!requireNamespace('TabulaMurisSenisData', quietly = TRUE)) {
BiocManager::install("TabulaMurisSenisData")
}
library(TabulaMurisSenisData)
library(SingleCellExperiment)
library(patchwork)
library(spacexr)
library(pbapply)Load single-cell data (Allen Brain atlas) and Visium data (mouse brain tissue section).
Load data
We have access to 23 annotated cell types in the single-cell data.
DimPlot(se_allen, group.by = "subclass")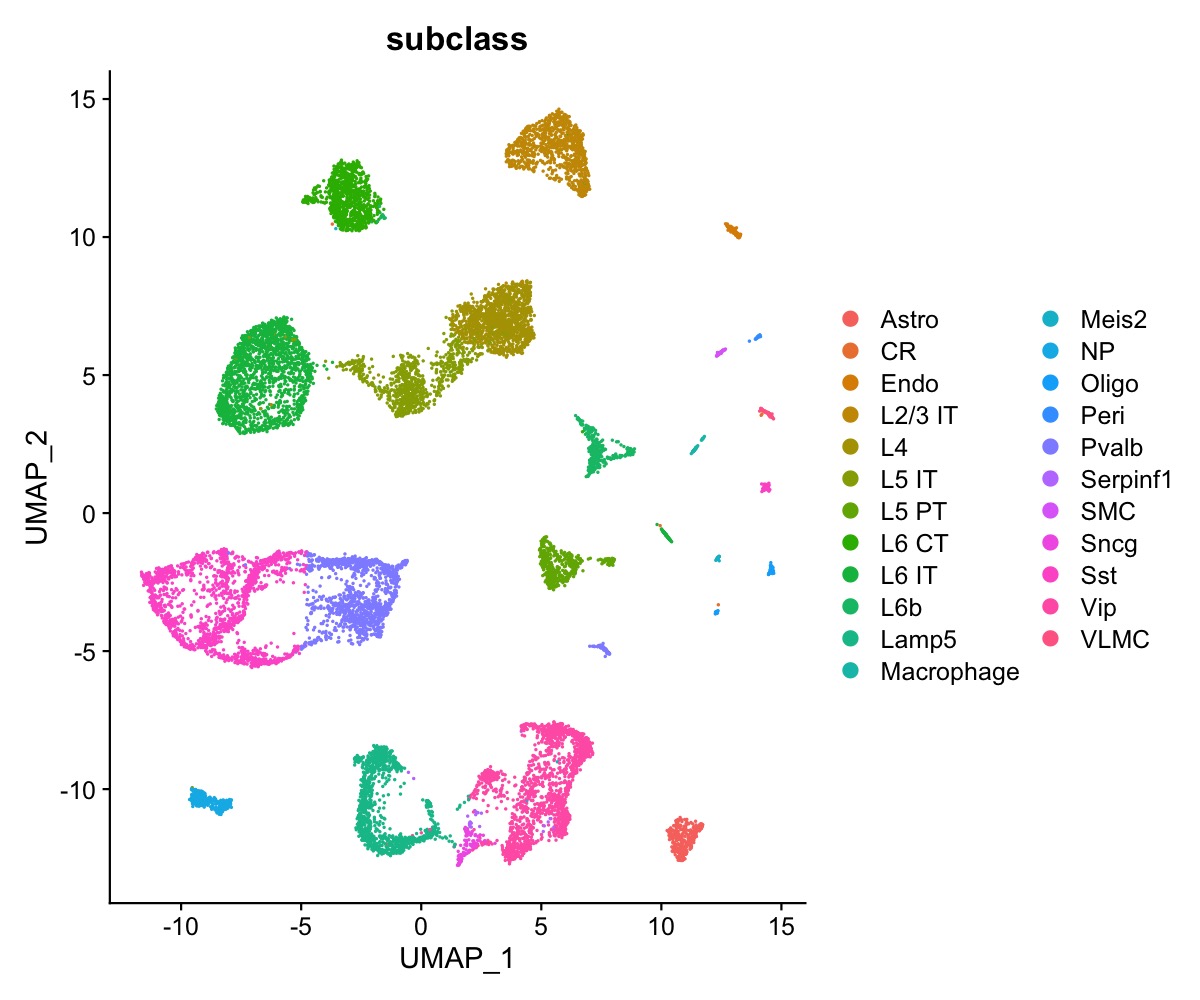
Create dummy expression profiles
Here we’ll create a synthetic Visium data set by sample single-cells from the Allen Brain atlas data set:
1. Downsample UMIs to 1%. This is to make sure that the synthetic data have roughly the same library sizes as the Visium mouse brain tissue section data set.
2. Calculate cell type weights. These weights will be used to determine the probability of a cell type being samples for the synthetic spots. Abundant cell type will be assigned higher probabilities and vice versa.
3. Sample cell numbers per synthetic spot. Here we’ll draw counts from a poisson distribution with the lambda (mean) parameter set to 10.
4. Select cell indices. The indices will be randomly selected using the cell type weights defined in step 2.
5. Aggregate expression vectors for selected indices. Each synthetic spot will consist of the averaged gene expression vectors obtained from the sampled cells. 6. Combine aggregated expression profiles to a final synthetic count matrix.
se_allen <- FindVariableFeatures(se_allen, nfeatures = 5e3)
se_allen <- se_allen[VariableFeatures(se_allen), ]
umis <- GetAssayData(se_allen, slot = "counts")
# 1. Down sample UMIs
bins <- split(1:ncol(se_allen), cut(1:ncol(se_allen), breaks = 50))
batches <- pblapply(bins, function(inds) {
umis_subset <- scuttle::downsampleMatrix(umis[, inds], prop = 0.01)
return(umis_subset)
}, cl = 7)
downsampled_umis <- do.call(cbind, batches)
# 2. Calculate cell type weights
props <- table(se_allen$subclass)
props <- props/sum(props) |> as.numeric()
# 3. Sample cells
# Set the average count
average_count <- 10
# Set the number of samples
num_samples <- 1e4
# Generate samples from a Poisson distribution with the specified average
set.seed(123)
sampled_counts <- rpois(num_samples, lambda = average_count)
sampled_counts[sampled_counts == 0] <- 1
# 4. Select cell indices
label_matrix <- sapply(names(props), function(label) se_allen$subclass == label)
sampled_indices <- pblapply(1:length(sampled_counts), function(i) {
x <- sample(x = names(props), size = sampled_counts[i], prob = props, replace = TRUE)
y <- table(x)
inds <- c()
for (lbl in unique(x)) {
inds <- c(inds, sample(x = which(label_matrix[, lbl]), size = y[lbl]))
}
return(inds)
}, cl = 7)
# 5. Aggregate expression vectors
bins <- split(1:num_samples, cut(1:num_samples, breaks = 50))
batches <- pblapply(bins, function(inds) {
umis_subset <- downsampled_umis[, sampled_indices[inds] |> unlist()]
umis_subset <- rowsum(umis_subset |> t(), group = rep(x = inds, sampled_indices[inds] |> sapply(length))) |> t()
return(umis_subset)
}, cl = 7)
# 6. Combine aggregated expression profiles
mini_bulk_umis <- do.call(cbind, batches)Export Allen brain atlas data for cell2location and stereoscope
Here we’ll export a subset of the single-cell data including 250 randomly selected cells per cell type. If a cell type has less than 250 cells, all cells will be selected. Only cell types with more than 10 cells are kept.
The exported matrices and tables will be used to run stereoscope and cell2location in a separate environment.
seed = 1337L
nCells_per_group <- 250
set.seed(seed)
# Sample barcodes
barcodes <- tibble(barcode = colnames(se_allen), group = se_allen$subclass) |>
group_by(group) |>
slice(sample(min(nCells_per_group, n())))
# Remove low abundant cell types
cells_per_celltype <- table(barcodes$group)
keep <- names(cells_per_celltype)[cells_per_celltype > 10]
barcodes <- barcodes |> filter(group %in% keep)
# Export matrices and tables
dir.create("synthetic_spots")
data.table::fwrite(GetAssayData(se_allen, slot = "counts")[, barcodes$barcode] |> t() |> as.data.frame(), file = "synthetic_spots/allen_brain_umis.tsv", quote = FALSE, sep = "\t", row.names = TRUE, col.names = TRUE)
writeLines(rownames(se_allen), con = "synthetic_spots/variable_genes.txt")
data.table::fwrite(se_allen@meta.data[barcodes$barcode, ] |> select(subclass), file = "synthetic_spots/metadata.tsv", sep = "\t", quote = FALSE, row.names = TRUE, col.names = TRUE)
data.table::fwrite(GetAssayData(se_Visium_agg, slot = "counts") |> t() |> as.data.frame(), file = "synthetic_spots/Visium_synthetic_spots_umis.tsv", quote = FALSE, sep = "\t", row.names = TRUE, col.names = TRUE)Create a Seurat object from the synthetic count matrix
Each synthetic spot will be named “barcode1”, barcode2”, …
For downstream steps, we need to normalize the data and select a set of top 5,000 variable genes.
colnames(mini_bulk_umis) <- paste0("barcode", 1:num_samples)
se_Visium_agg <- CreateSeuratObject(counts = mini_bulk_umis, assay = "Spatial")
se_Visium_agg <- se_Visium_agg |>
NormalizeData() |>
FindVariableFeatures(nfeatures = 5000)Run NNLS
The RunNNLS() method requires a Seurat
object with normalized 10x Visium data and a Seurat object
with normalized single-cell data. The groups argument
defines what meta data column the cell type labels should be taken from
in the single-cell Seurat object. In our single-cell
Seurat object, the labels are stored in the “subclass”
column.
Computation time
For the NNLS method, we’ll run 10 rounds, adding 10,000 spots to each
round and starting with 10,000 spots. Below we create a “large” Seurat
object with 100,000 spots by merging our se_Visium_agg
object 10 times.
# Merge Seurat object to have 100k spots
se_Visium_agg_large <- merge(se_Visium_agg, y = list(se_Visium_agg, se_Visium_agg, se_Visium_agg, se_Visium_agg, se_Visium_agg, se_Visium_agg, se_Visium_agg, se_Visium_agg, se_Visium_agg))
DefaultAssay(se_Visium_agg_large) <- "Spatial"
se_Visium_agg_large <- FindVariableFeatures(se_Visium_agg_large, nfeatures = 5e3)Since the NNLS method runs in a matter of seconds, we’ll run each round in 10 iterations to compute average computation times.
nnls_results <- list()
for (i in seq(1e4, 1e5, 1e4)) {
print(i)
DefaultAssay(se_Visium_agg_large) <- "Spatial"
se_Visium_agg_subset <- se_Visium_agg_large[, 1:i]
iters <- sapply(1:10, function(n) {
cat(paste0(" iter: ", n, "\n"))
ti <- Sys.time()
tmp <- RunNNLS(object = se_Visium_agg_subset, nCells_per_group = 250,
singlecell_object = se_allen,
groups = "subclass", verbose = FALSE)
stamp <- Sys.time() - ti
return(stamp)
})
nnls_results[[paste0("n", i)]] <- tibble(n = i, stamp = iters)
rm(se_Visium_agg_subset)
}
nnls_stamps <- do.call(bind_rows, nnls_results)Now we can plot the average computation times for each data set size.
p <- ggplot(nnls_stamps |> group_by(n) |> summarize(mean = mean(stamp), sd = sd(stamp)),
aes(x = n, y = mean, ymin = mean - sd, ymax = mean + sd)) +
geom_errorbar(width = 0) +
geom_point() +
labs(x = "Number of spots", y = "Seconds", title = "Computation time with NNLS (5,000 genes)") +
theme_minimal() +
scale_x_continuous(labels=function(x) format(x, big.mark = ",", scientific = FALSE),
breaks = seq(1e4, 1e5, 1e4)) +
theme(axis.text.x = element_text(angle = 50, hjust = 1), panel.border = element_rect(fill = NA, colour = "black"))
p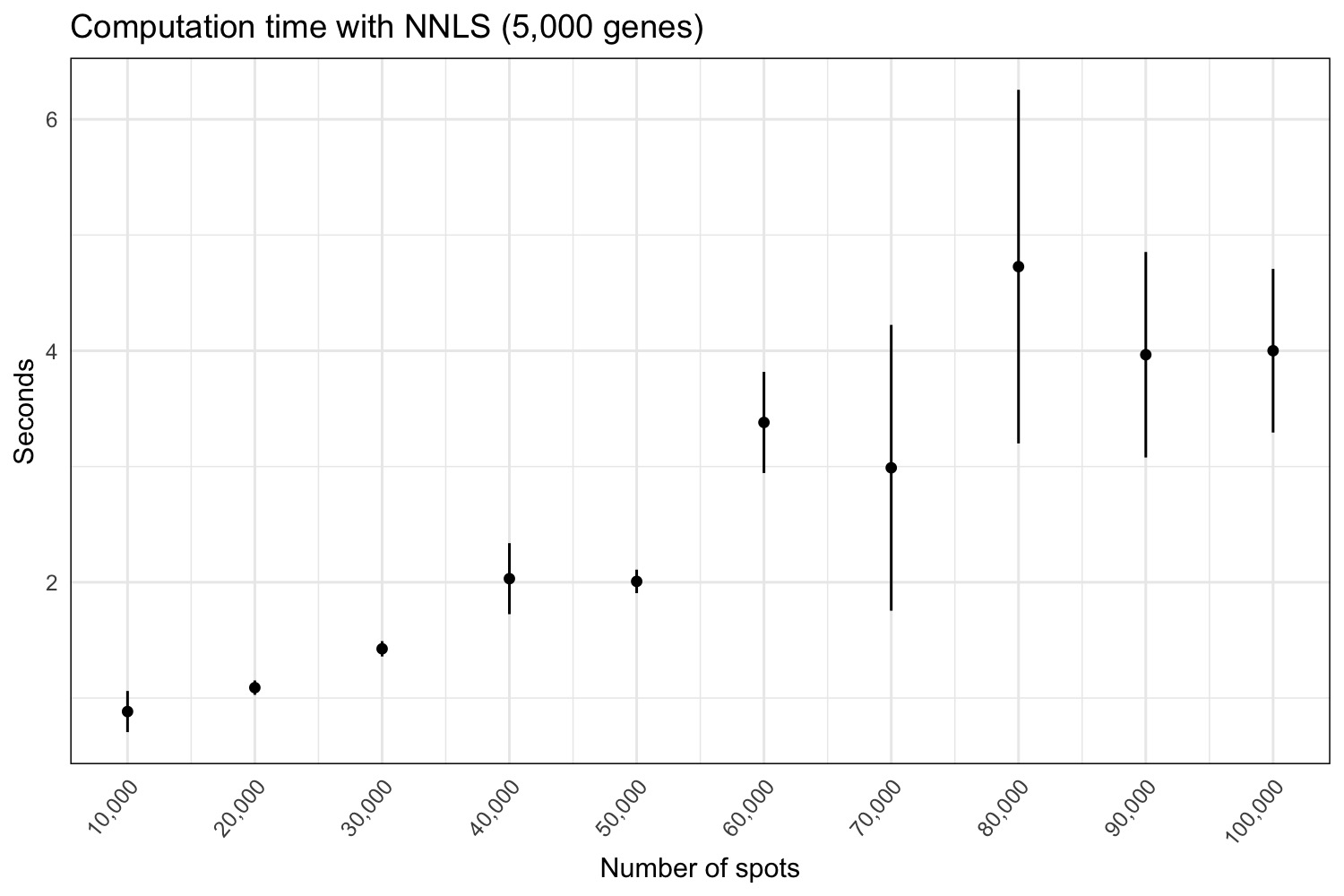
Compute performance metrics
For the performance assessment, we’ll run the NNLS method on our
10,000 spots se_Visium_agg data set. We can get the
expected counts by counting the number of cell types using our sampled
indices.
# Calculate expected counts per spot
expected_counts <- do.call(bind_rows, pblapply(seq_along(sampled_indices), function(i) {
x <- se_allen$subclass[sampled_indices[[i]]] |> table()
tibble(spot = paste0("barcode", i), celltype = names(x), lbl = x |> as.integer())
}, cl = 7))
# Run NNLS for 1e4 spots
DefaultAssay(se_Visium_agg) <- "Spatial"
se_Visium_agg <- RunNNLS(object = se_Visium_agg,
singlecell_object = se_allen,
groups = "subclass", verbose = FALSE, min_prop = 0)
# cast to wide format
props <- expected_counts |> tidyr::pivot_wider(id_cols = spot, names_from = celltype, values_from = lbl) |>
tibble::column_to_rownames(var = "spot") |>
as.matrix()
props[is.na(props)] <- 0
props <- prop.table(props, margin = 1)
props <- props[, colnames(props) != "CR"]
props <- props[, rownames(se_Visium_agg)]
# Compare proportions
pred_all <- tibble(inf_props_nnls = GetAssayData(se_Visium_agg, slot = "data") |>
as.matrix() |> t() |> as.numeric(),
exp_props = props |> as.numeric(),
celltype = rep(rownames(se_Visium_agg), each = 1e4))Run RCTD
Now we can run the RCTD method on our 10,000 spots data set and time the computation. The RCTD method could not be run locally for larger data sets and we therefore only ran the deconvolution for 10,00 spots. (Took ~19 minutes to run on a Macbook Pro 2017, 3.1 GHz Quad-Core Intel Core i7, 16GB).
# set up reference
Idents(se_allen) <- "subclass"
# extract information to pass to the RCTD Reference function
celltypes <- gsub(pattern = "\\/", replacement = ".", x = se_allen$subclass)
celltypes <- celltypes[celltypes != "CR"]
cluster <- as.factor(gsub(pattern = "\\/", replacement = ".", x = celltypes))
nUMI <- colSums(GetAssayData(se_allen, slot = "counts"))
reference <- Reference(GetAssayData(se_allen, slot = "counts")[, barcodes$barcode], cluster[barcodes$barcode], nUMI[barcodes$barcode])
# set up query with the RCTD function SpatialRNA
colnames(mini_bulk_umis) <- paste0("barcode", 1:ncol(mini_bulk_umis))
coords <- data.frame(x = 1:1e4, y = 1:1e4, row.names = paste0("barcode", 1:1e4))
query <- SpatialRNA(coords = coords, counts = mini_bulk_umis, nUMI = colSums(mini_bulk_umis))
# Run RCTD
ti <- Sys.time()
RCTD <- create.RCTD(query, reference, max_cores = 7)
RCTD <- run.RCTD(RCTD, doublet_mode = 'full')
rctd_stamp <- Sys.time() - tiCompute performance metrics
The results of RCTD full mode are stored in
@results$weights. To obtain proportion estimates, we
normalize the weights using normalize_weights so that they
sum to one. Each entry represents the estimated proportion of each cell
type on each spot.
barcodes <- colnames(RCTD@spatialRNA@counts)
weights <- RCTD@results$weights
norm_weights <- normalize_weights(weights)
colnames(norm_weights) <- gsub(pattern = "\\.", replacement = "/", x = colnames(norm_weights))
# Compare proportions
pred_all$inf_props_rctd <- norm_weights[, colnames(props)] |> as.numeric()Seurat
Finally, we’ll run the label transfer method available from
Seurat. The method returns prediction scores for each cell
type and spot which cannot be directly interpreted as proportions.
However, if we normalize the prediction scores we can use them as a
proxy for cell type proportions to calculate our performance
metrics.
Computation time
The Seurat method runs relatively fast and we therefore logged the computation time running on synthetic Visium data from 10,000 to 100,000 spots.
se_Visium_agg_large <- SetIdent(se_Visium_agg_large, value = "Spatial")
se_Visium_agg_large <- se_Visium_agg_large |> ScaleData() |> RunPCA()
seurat_results <- list()
for (i in seq(1e4, 1e5, 1e4)) {
print(i)
DefaultAssay(se_Visium_agg_large) <- "Spatial"
se_Visium_agg_subset <- se_Visium_agg_large[, 1:i]
ti <- Sys.time()
anchors <- FindTransferAnchors(reference = se_allen,
query = se_Visium_agg_subset)
predictions.assay <- TransferData(anchorset = anchors,
refdata = se_allen$subclass,
prediction.assay = TRUE,
weight.reduction = se_Visium_agg_subset[["pca"]],
dims = 1:30)
stamp <- Sys.time() - ti
seurat_results[[paste0("n", i)]] <- tibble(n = i, stamp = stamp)
}
seurat_stamps <- do.call(bind_rows, seurat_results)Below we compare the run time for the NNLS and Seurat methods.
gg <- bind_rows(nnls_stamps |> mutate(method = "nnls") |> mutate(stamp = as.numeric(stamp)),
seurat_stamps |> mutate(method = "Seurat") |> mutate(stamp = as.numeric(stamp)*60))
p <- ggplot(gg |> group_by(n, method) |> summarize(mean = mean(stamp), sd = sd(stamp)),
aes(x = n, y = mean, ymin = mean - sd, ymax = mean + sd, color = method)) +
geom_point() +
geom_line() +
labs(x = "Number of spots", y = "Seconds", title = "Computation time with NNLS and Seurat (5,000 genes)") +
theme_minimal() +
scale_x_continuous(labels=function(x) format(x, big.mark = ",", scientific = FALSE),
breaks = seq(1e4, 1e5, 1e4)) +
scale_y_log10() +
annotation_logticks() +
theme(axis.text.x = element_text(angle = 50, hjust = 1), panel.border = element_rect(fill = NA, colour = "black"))
p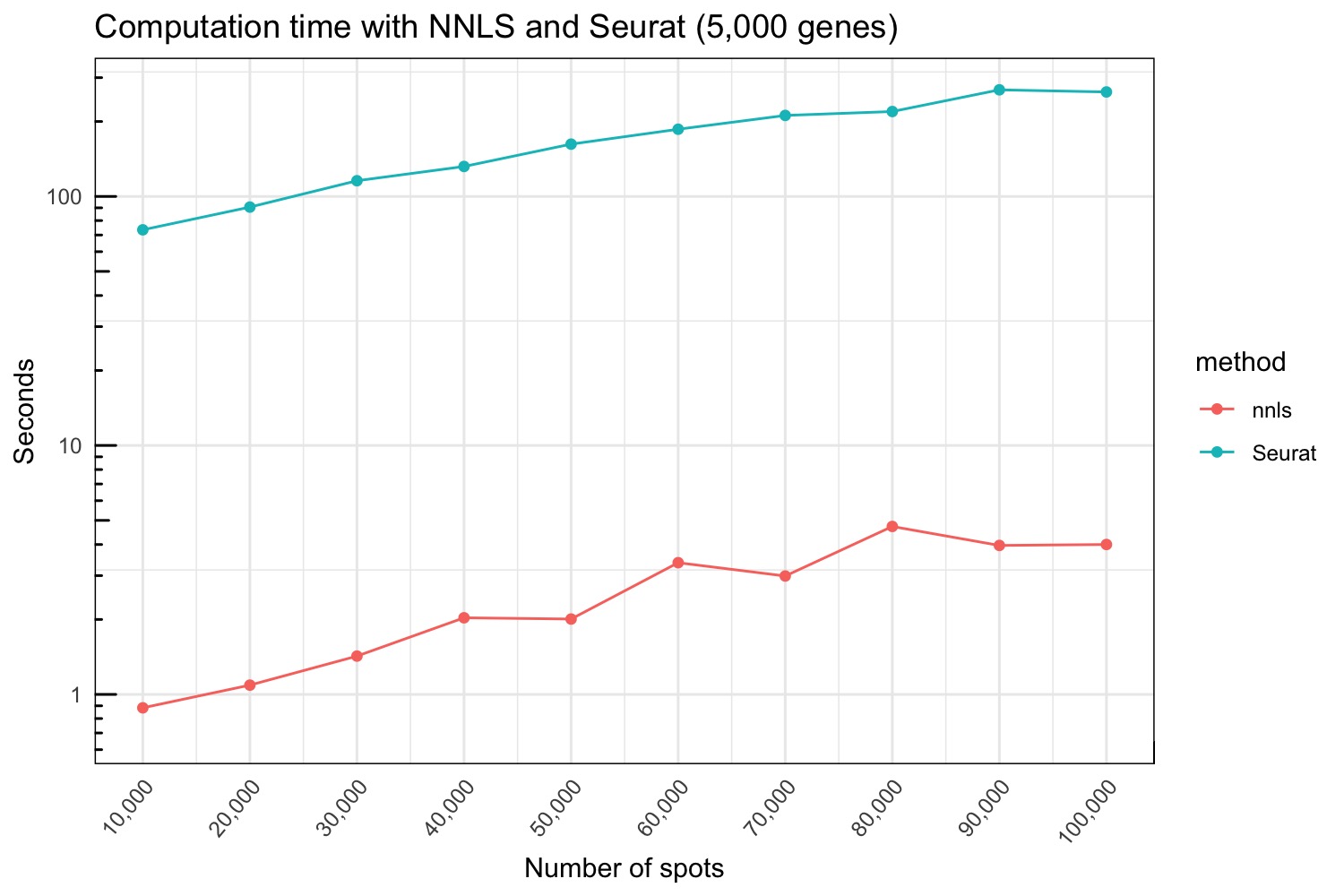
Compute performance metrics
Again, we can run the Seurat method on our 10,000 spots data set and normalize the results scores to obtain proportion estimates and calculate performance metrics.
se_Visium_agg <- SetIdent(se_Visium_agg, value = "Spatial")
se_Visium_agg <- se_Visium_agg |> ScaleData() |> RunPCA()
anchors <- FindTransferAnchors(reference = se_allen[, se_allen$subclass != "CR"],
query = se_Visium_agg)
predictions.assay <- TransferData(anchorset = anchors,
refdata = se_allen$subclass[se_allen$subclass != "CR"],
prediction.assay = TRUE,
weight.reduction = se_Visium_agg[["pca"]],
dims = 1:30)
predictions.assay.props <- prop.table(predictions.assay@data[rownames(predictions.assay) != "max", ], margin = 2)
# Compare proportions
pred_all$inf_props_seurat <- predictions.assay.props[rownames(se_Visium_agg@assays$celltypeprops), ] |> t() |> as.numeric()Stereoscope
For deconvolution with stereoscope, we used the scvi implementation, following the tutorial ‘STereoscope applied to left ventricule data’. First, a model was trained on the scRNA-seq data with 1,000 epochs. Next, the model was used to deconvolve the synthetic Visium expression profiles in 2,000 epochs. For the stereoscope deconvolution, we used a NVIDIA A100-SMX4-80GB Tensor core GPU.
Compute performance metrics
stereoscope <- read.table("synthetic_spots/stereoscope/W.2023-08-17102550.170200.tsv", sep = "\t", header = TRUE, row.names = 1, check.names = FALSE)
pred_all$inf_props_stereoscope <- stereoscope[, rownames(se_Visium_agg@assays$celltypeprops)] |> as.matrix() |> as.numeric()Cell2location
For deconvolution with cell2location, we followed the tutorial ‘Mapping human lymph node cell types to 10X Visium with Cell2location’. First, a model was trained on the scRNA-seq data with 1,000 epochs. Next, the model was used to deconvolve the synthetic Visium expression profiles in 30,000 epochs. For the cell2location deconvolution, we used a NVIDIA A100-SMX4-80GB Tensor core GPU.
Compute performance metrics
Load cell2location cell abundances.
cell2location <- read.table("synthetic_spots/cell2location/q05_cell_abundance_w_sf.csv", sep = ",", header = TRUE, row.names = 1, check.names = FALSE)
cell2location_props <- prop.table(cell2location |> as.matrix(), margin = 1)
colnames(cell2location_props) <- gsub(pattern = "q05cell_abundance_w_sf_", replacement = "", x = colnames(cell2location_props))
pred_all$inf_props_cell2location <- cell2location_props[, rownames(se_Visium_agg@assays$celltypeprops)] |> as.matrix() |> as.numeric()Run time for 10k spots
p <- ggplot(run_time, aes(method, time)) +
geom_col() +
scale_y_log10() +
geom_label(aes(method, time, label = time_format)) +
labs(x = "Deconvolution method", y = "time in seconds", title = "Run time for 10,000 spots")
p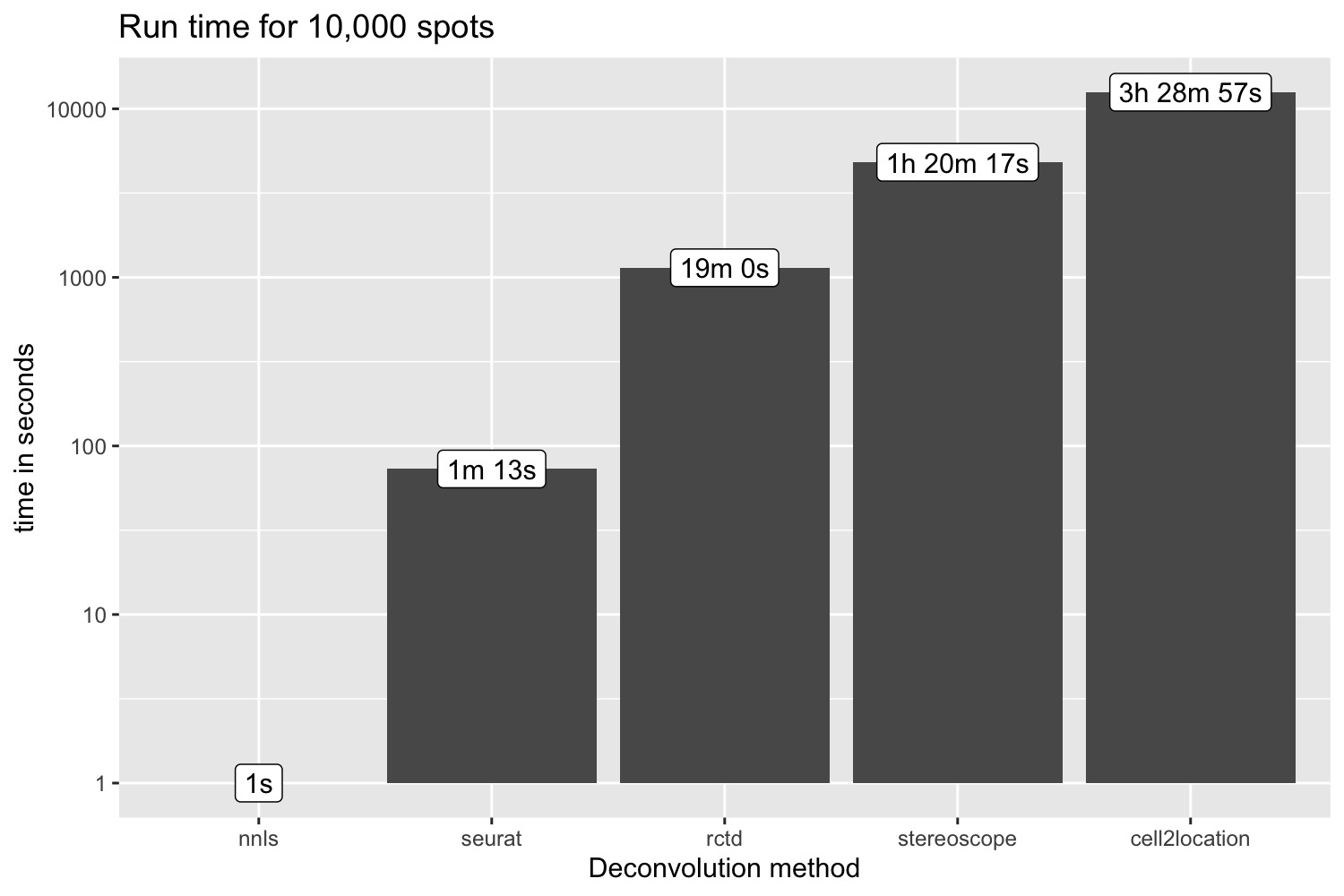
pred_all_long <- pred_all |>
tidyr::pivot_longer(cols = c("inf_props_nnls", "inf_props_rctd", "inf_props_seurat",
"inf_props_stereoscope", "inf_props_cell2location"),
names_to = "method", values_to = "inf_props") |>
mutate(method = gsub(pattern = "inf_props_", replacement = "", x = method)) |>
mutate(method = factor(method, levels = c("nnls", "rctd", "stereoscope", "cell2location", "seurat"))) |>
group_by(celltype, method) |>
summarize(cor = cor(exp_props, inf_props),
rmse = rmse(exp_props, inf_props), .groups = "drop")
p1 <- ggplot(pred_all_long, aes(method, cor)) +
geom_violin(width = 1) +
labs(title = "Pearson correlation (inferred vs expected proportions)")
p2 <- ggplot(pred_all_long, aes(method, rmse)) +
geom_violin(width = 1) +
labs(title = "RMSE (inferred vs expected proportions)")
p1 + p2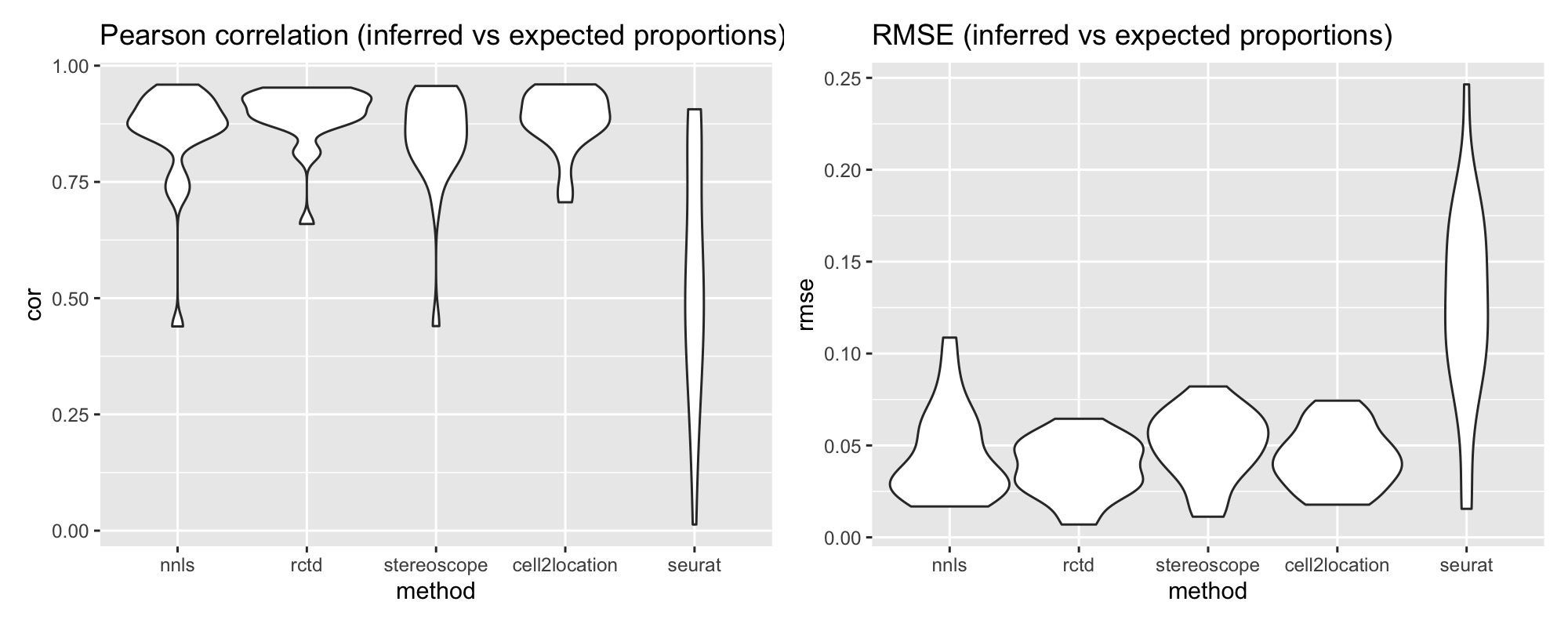
Scatter plots
pred_all_long <- pred_all |> tidyr::pivot_longer(cols = c("inf_props_nnls", "inf_props_rctd", "inf_props_seurat",
"inf_props_stereoscope", "inf_props_cell2location"),
names_to = "method", values_to = "inf_props") |>
mutate(method = gsub(pattern = "inf_props_", replacement = "", x = method))
p <- ggplot(pred_all_long,
aes(exp_props, inf_props)) +
geom_point(size = 0.5) +
facet_grid(celltype~method) +
geom_smooth(method = lm) +
scale_x_continuous(limits = c(0, 1)) +
scale_y_continuous(limits = c(0, 1)) +
geom_abline(slope = 1, intercept = 0, linetype = "dashed", color = "red") +
labs(x = "Expected proportions", y = "Inferred proportions") +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5))
p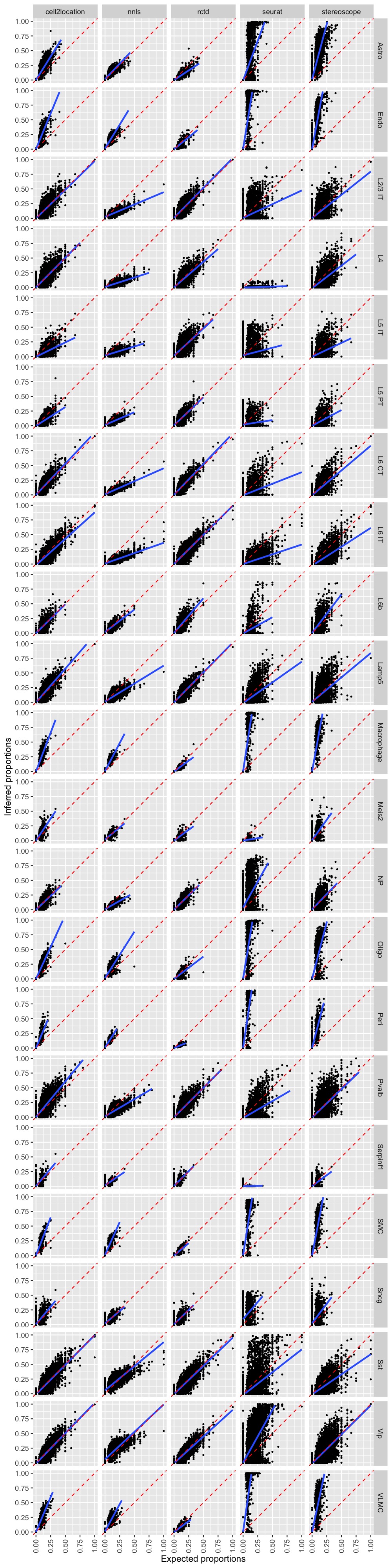
Package versions
semla: 1.4.1RcppML: 0.5.6
Session info
## R version 4.4.2 (2024-10-31)
## Platform: aarch64-apple-darwin20.0.0
## Running under: macOS Sequoia 15.7.3
##
## Matrix products: default
## BLAS/LAPACK: /Users/javierescudero/miniconda3/envs/r-semlaupd/lib/libopenblas.0.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: Europe/Stockholm
## tzcode source: system (macOS)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## loaded via a namespace (and not attached):
## [1] digest_0.6.37 desc_1.4.3 R6_2.6.1 fastmap_1.2.0
## [5] xfun_0.53 cachem_1.1.0 knitr_1.50 htmltools_0.5.8.1
## [9] rmarkdown_2.29 lifecycle_1.0.4 cli_3.6.4 sass_0.4.9
## [13] pkgdown_2.1.1 textshaping_0.4.0 jquerylib_0.1.4 systemfonts_1.3.1
## [17] compiler_4.4.2 rstudioapi_0.17.1 tools_4.4.2 ragg_1.3.3
## [21] bslib_0.9.0 evaluate_1.0.5 yaml_2.3.10 jsonlite_1.9.0
## [25] rlang_1.1.5 fs_1.6.5 htmlwidgets_1.6.4